Ciliopathy & Eye diseases
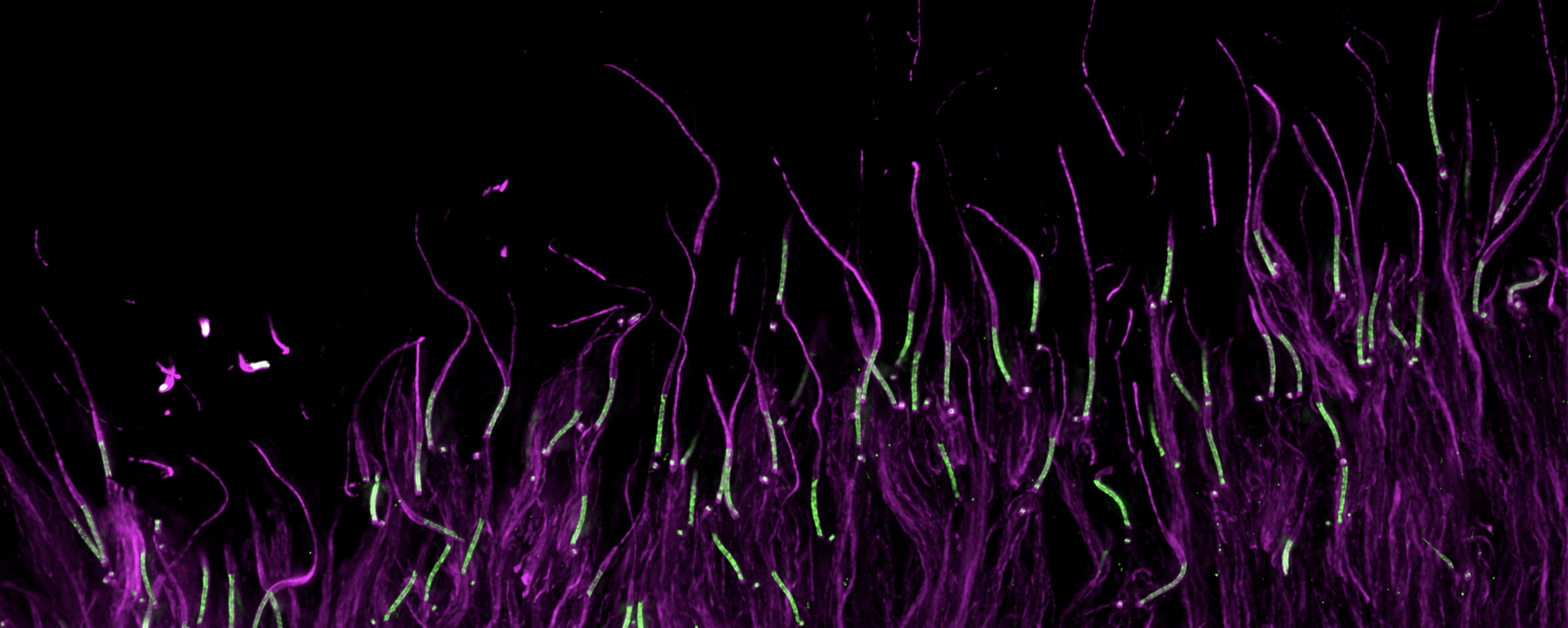
Image: Mouse photoreceptors cells in U-ExM (Olivier Mercey)
Retinitis pigmentosa (RP) is a group of genetic eye disorders that primarily affect the retina, which is the light-sensitive tissue lining the back of the eye. RP is characterized by progressive degeneration of the retina's photoreceptor cells, namely the rods and cones, which are responsible for capturing and processing light to send visual signals to the brain. As these cells deteriorate over time, individuals with RP experience a gradual loss of vision, typically starting with night blindness and then progressing to tunnel vision and eventually, in some cases, complete blindness.
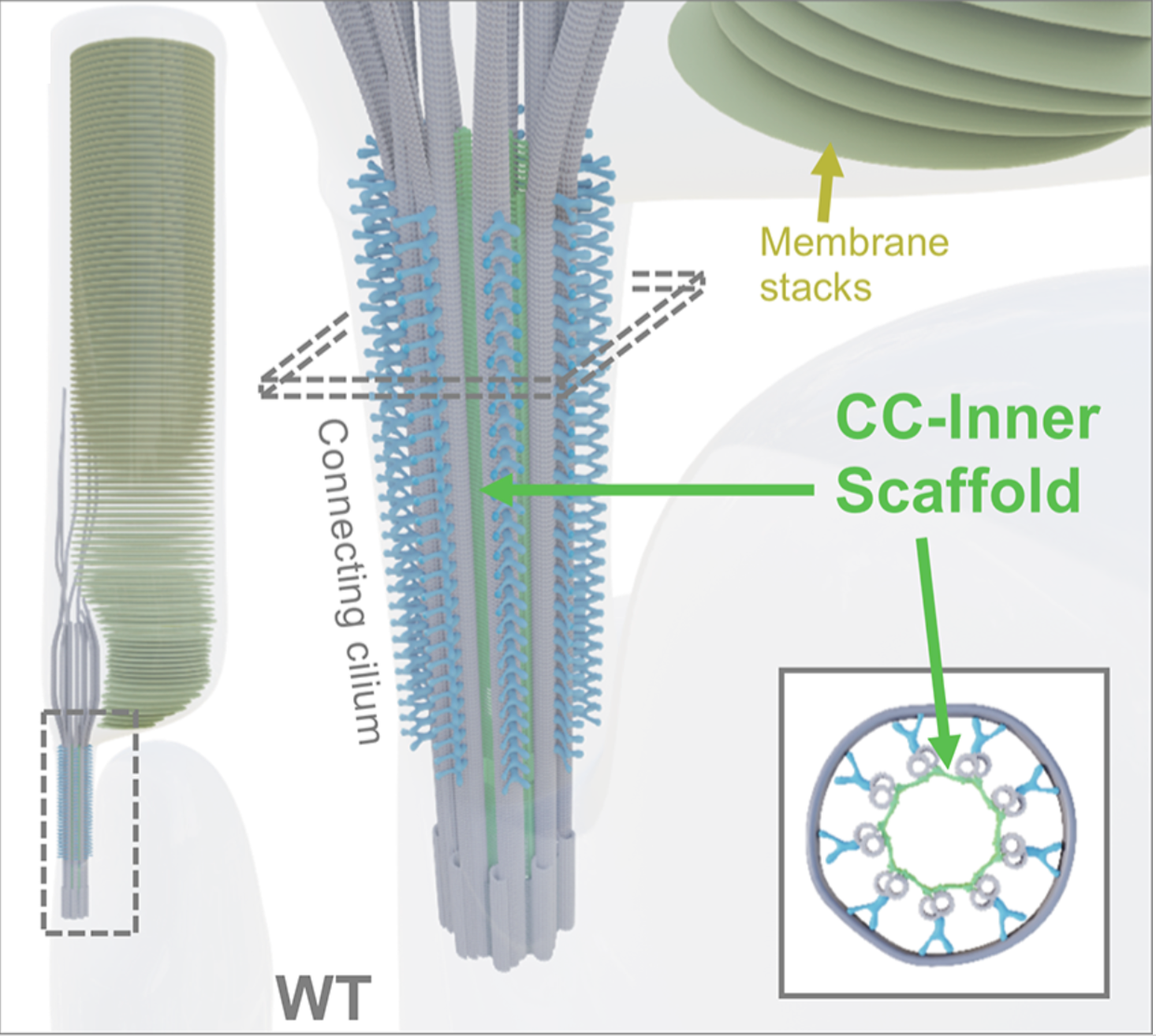
The laboratory is especially focused on a specific organelle within the photoreceptor known as the connecting cilium. This structure exhibits numerous structural similarities with the centriole, notably featuring an inner scaffold comprised partially of the POC5, FAM161A, and Centrin-2 proteins, all of which are implicated in retinitis pigmentosa. Our research aims to gain a deeper comprehension of the structure and functionality of the connecting cilium to enhance our understanding of the underlying causes of these diseases.
 Retinitis PIgmentosa 28 (RP28) associated wit the gene FAM161A
Retinitis PIgmentosa 28 (RP28) associated wit the gene FAM161A
In collaboration with the groups of Dr. Corinne Kostic and Prof. Yvan Arsenijevic (Hôpital Ophtalmique Jules-Gonin and UNIL), we use expansion microscopy (U-ExM), electron microscopy and cell biology approaches to understand the molecular mechanisms that lead to retinal degeneration. In particular, our research focused on the human FAM161A gene whose mutations have been linked to autosomal recessive RPs. Using U-ExM in mouse retina, we recently showed that Fam161a disruption leads to the loss of a sub-cellular structure specific to the photoreceptors cells, the inner scaffold of the connecting cilium. The loss of this scaffold triggers microtubule doublet spreading, prior to photoreceptor degeneration.
Link to the article : https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.3001649
 Leber congenital amaurosis associated with the gene LCA5
Leber congenital amaurosis associated with the gene LCA5
Leber congenital amaurosis (LCA) is a rare and severe genetic eye disorder that is present from birth. It is one of the most common causes of congenital blindness in infants and young children. LCA primarily affects the retina, the light-sensitive tissue at the back of the eye, and it typically results in severe visual impairment or blindness early in life. In collaboration with the group of Ronald Roepman, we reveal the structural and molecular defects underlying LCA type 5 (LCA5) with nanoscale resolution using U-ExM. Moreover we used expansion microscopy for the first time to assess retinal (gene) therapy efficacy at the molecular level.
Link to the article: https://insight.jci.org/articles/view/169162
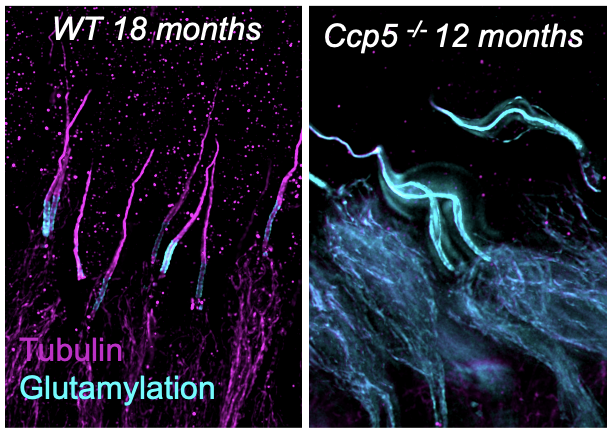 Retinitis Pigmentosa associated witH the gene AGBL5
Retinitis Pigmentosa associated witH the gene AGBL5