Research projects
Oncogene-induced DNA replication stress
The long-term goal of our research is to obtain a better understanding of cancer, such that effective, non-toxic therapies can be developed. Towards this goal, we aim to identify differences between normal cells and cancer cells. Our laboratory, in collaboration with Vassilis Gorgoulis (University of Athens Medical School) and, independently, the laboratory of Jiri Bartek (Danish Cancer Society) reported that human cancers are characterized by the presence of DNA replication stress (Figure 1). Importantly, normal cells, even from highly replicating tissues, do not have DNA replication stress.
DNA replication stress leads to damaged replication forks and formation of DNA double-strand breaks. Thus, replication stress can explain the high degree of genomic instability that is present in human cancers, as well as why the p53 gene is the most frequently mutated gene in cancer. DNA damage activates p53, which then induces cell death or senescence. Thus, in the face of DNA replication stress, cancer cells can proliferate rapidly only after the p53 gene has been mutated. Our studies have further identified activated oncogenes as the cause of DNA replication stress in human cancers. Interestingly, it appears that most oncogenes induce DNA replication stress.
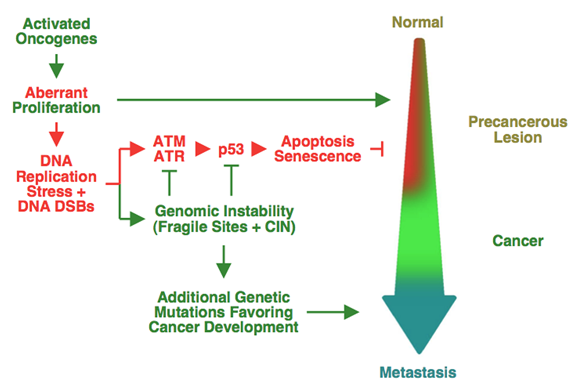
Figure 1. Model for oncogene-induced DNA replication stress in human cancers selecting for p53 mutations and driving genomic instability
Mechanism by which activated oncogenes induce DNA replication stress
We recently proposed a mechanism to explain how activated oncogenes induce DNA replication stress (Figure 2 and Movie 1). We found that both normal and cancer cells establish a large number of DNA replication origins, from which DNA replication may initiate. In normal cells, the origins that are located in genes are inactivated by the transcription machinery during the G1 phase of the cell cycle. The inactivation is very efficient, because the G1 phase lasts for ten hours or more. As a result, when cells enter S phase, DNA replication begins only from origins that are located between genes. However, in cancer cells, the G1 phase is shortened and there is not enough time for transcription to inactivate the replication origins within large genes. Initiation of replication from these origins, leads to collisions between the replication and transcription machinery and DNA damage.
The shortening of the length of the G1 phase of the cell cycle in cancer cells is due to the activation of oncogenes, which drive entry of the cancer cells into the cell cycle. Thus, the presence of DNA replication stress in human cancers can be viewed as a necessary side-effect of oncogene activation.
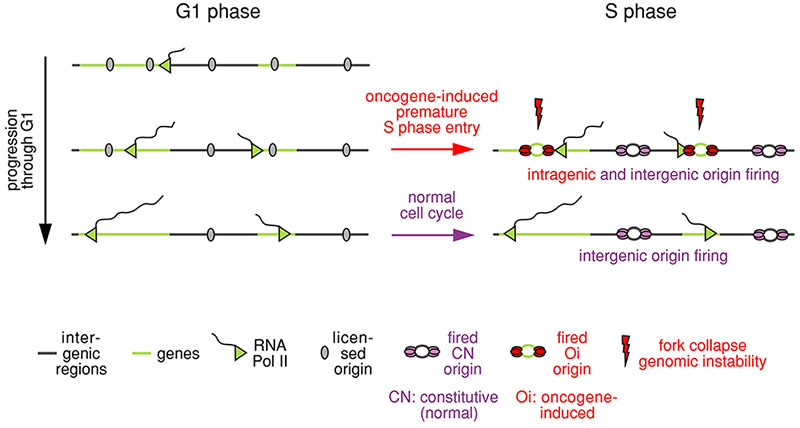
Figure 2. Mechanism to explain how oncogenes induce DNA replication stress
Shortening of the length of the G1 phase of the cell cycle by activated oncogenes leads to firing of DNA replication within genes, collisions between the replication and transcription machineries, DNA breaks and genomic instability.
Movie 1. Mechanism to explain how oncogenes induce DNA replication stress
The DNA corresponding to a gene is shown in red; the DNA replication machinery is in green; and the transcription machinery in yellow.
Oncogene-induced DNA replication stress as a therapeutic target
Since the presence of DNA replication stress distinguishes cancer cells from normal cells, it could serve as a target for cancer-specific therapies. Targeting DNA replication stress has the advantage that cancer cells cannot develop resistance by switching their growth dependency from one activated oncogene to another, since most oncogenes induce replication stress.
We envision two broad approaches that could help the development of novel therapies. First, we could exploit our understanding of how oncogenes induce DNA replication stress to enhance the level of replication stress present in cancer cells. Second, since DNA replication stress leads to damaged replication forks, we could inhibit the relevant DNA repair pathways. We have identified break-induced replication (BIR) as a major pathway for repair of collapsed forks in cancer cells. BIR relies on homologous recombination to restart DNA replication from collapsed forks. BIR-initiated forks retain the D-loop formed during homologous recombination and replicate DNA in a conservative manner (Figure 3). It is very likely that the differences between BIR-initiated and origin-initiated forks could be exploited for the development of cancer therapies. We have already identified genes that function in BIR, for example, RAD52, POLD3 and POLD4, and shown that suppressing these genes inhibits proliferation specifically of cancer cells.
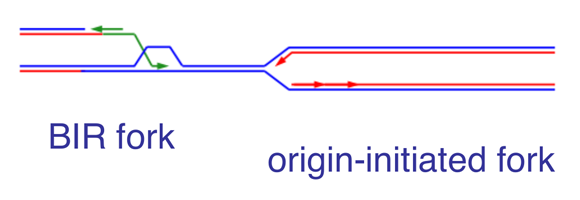
Figure 3. Comparison of BIR-initiated and origin-initiated forks
BIR-initiated forks retain the D-loop formed during strand invasion and replicate DNA in a conservative manner.
Methodology
Research in the laboratory relies heavily on high throughput sequencing approaches to study DNA replication (Figure 4). We also employ siRNA screens to identify genes that function in the response to DNA replication stress and use both cell lines and organoids as model systems.
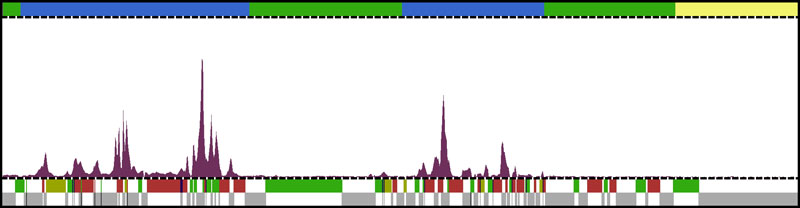
Figure 4. DNA replication origin firing in U2OS cells, as determined by the EdUseq method
The top bar indicates replication timing domains (early S, blue; mid S, green; late S, yellow). The lower bar indicates the genes (in color, based on the direction of transcription: green, forward; red, reverse) and intergenic regions (in gray).
Development of SARS-CoV-2 Inhibitors
The COVID-19 pandemic has already had significant impact on the health, life-style and economy of every society worldwide. One can argue that scientists competent to contribute to our understanding of this disease have a moral obligation to do so. Our expertise is best geared towards development of therapeutics. We have therefore initiated efforts to identify chemical compounds that inhibit replication of SARS-CoV-2, the agent responsible for COVID-19.
Mpro (3CLpro) is the main protease of SARS-CoV-2. Mpro cleaves the viral precursor proteins PP1a and PP1ab into functional viral proteins. The inhibition of Mpro would therefore hinder multiple processes essential for viral replication, as it would prevent the production of the mature forms of most viral proteins.
Two groups have recently published the results of in silico screens of more than one billion compounds targeting the Mpro protease. Our group was involved in one of these screens. Using the results generated by these two screens, we had more than one thousand chemical compounds synthesized and examined their ability to inhibit Mpro in vitro.
Remarkably, we identified five structurally related chemical compounds that bind to Mpro and inhibit its activity with IC50 values in the low micromolar range (Fig. 5; ref 1). These compounds are not potent enough to be used for treatment, but we are continuing our efforts to improve them. Even if our efforts may not affect the current pandemic, they have the potential to affect future pandemics, because the Mpro protease is very highly conserved in coronaviruses.
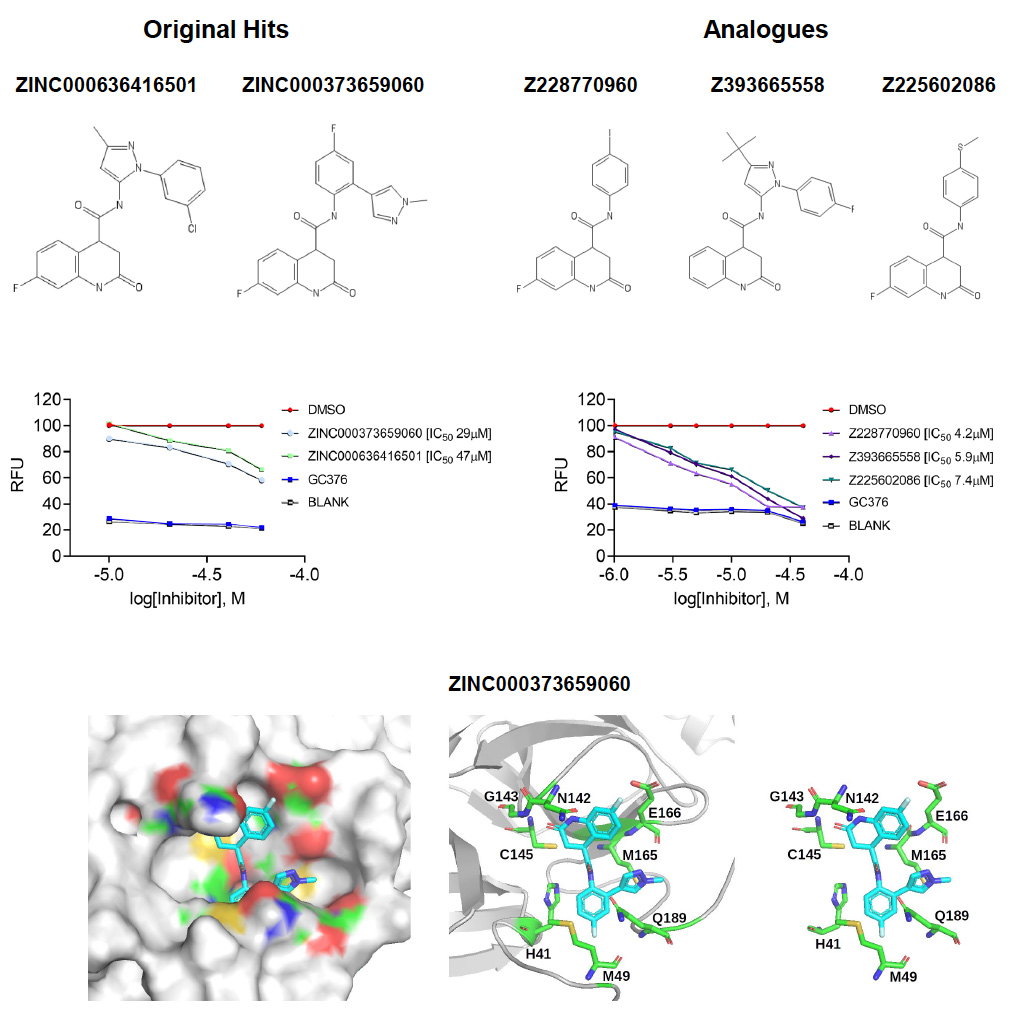
Figure 5. Low micromolar SARS-CoV-2 Mpro inhibitors
Top panels: Chemical structures of two hits identified by the in silico screens (left) and of three analogues of these hits (right) identified by biochemical screening.
Middle panels: Dose-response curves showing inhibition of Mpro by the identified compounds. RFU, relative fluorescence units. The lower the IC50 value, the more potent a compound is.
Lower panels: Model showing how one of the inhibitors binds to Mpro. The model is based on the in silico screen data of Ton et al (DOI: 10.1002/minf.202000028).
References
1. Rossetti GG, Ossorio M, Barriot S, Tropia L, Dionellis VS, Gorgulla C, Arthanari H, Mohr P, Gamboni R, Halazonetis TD. (2020). Identification of low micromolar SARS-CoV-2 Mpro inhibitors from hits identified by in silico screens. bioRxiv, DOI: 10.1101/2020.12.03.409441